
The workshop will start at 9AM on Monday, August 22, 2011 and finish at 5PM on Friday, August 26, 2011. Each of the five workshop days will consist of approximately 3 hours of lectures and 5 hours of practical exercises in the form of hands-on-tutorials on local workstations.
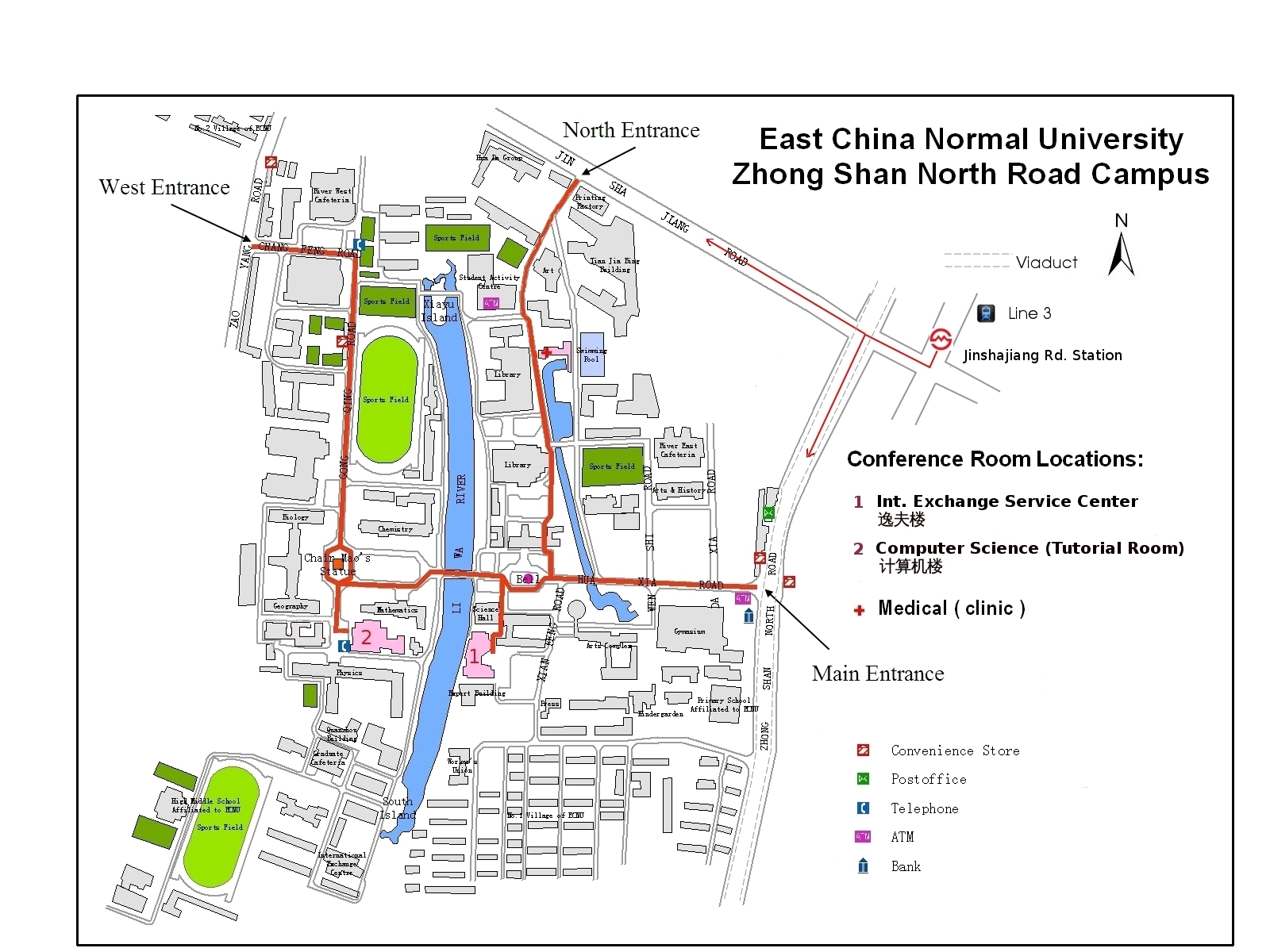
The workshop will take place in Room 200 of the ECNU Computer Science Building (click here for a campus map)
The official workshop language (including all course materials) will be English (limited support during the hands-on sessions can also be provided in Chinese).
Linux workstations with all required software will be provided for the hands-on sessions. Personal laptops are not required. While AmberTools is available for free, the AMBER software package is officially distributed by UCSF subject to a licensing agreement. The workshop will use version 11 of AMBER and version 1.5 of AmberTools.
Instructors: Ross Walker (RW), Andreas Goetz (AG)
Tutors: Ye Mei, (YM), Yun Xiang, (YX), Xingyu Wang (XW)
(L) = Lecture,
(H) = Hands on Tutorial
Day 1 (Monday 22nd Aug 2011)
| 9:00 - 9:45 | Registration. | |
| 9:45 - 10:00 | Welcome / Introduction to the Workshop. (L) | |
| 10:00 - 10:45 | Introduction to force fields and molecular dynamics. (Part 1) (L) | RW |
| 10:45 - 11:15 | Coffee break. | |
| 11:15 - 12:00 | Introduction to force fields and molecular dynamics. (Part 2) (L) | RW |
| 12:00 - 13:30 | Lunch. | |
| 13:30 - 14:15 | Overview of the AMBER package and its programs. (L) | RW |
| 14:15 - 17:00 | Hands-on session 1 - (Introduction to molecular dynamics simulations using AMBER) (H) | RW/AG/YM/YX/XW |
Day 2 (Tuesday 23rd Aug 2011)
| 8:30 - 10:00 | Hands-on session 2 - (Using VMD to visualize AMBER simulations) (H) | RW/AG/YM/YX/XW |
| 10:00 - 10:45 | Overview of AMBER Force Fields and Solvent Models. (L) | RW |
| 10:45 - 11:15 | Coffee break. | |
| 11:15 - 12:00 | Dealing with non-standard residues. (L) | RW |
| 12:00 - 13:30 | Lunch. | |
| 13:30 - 15:30 | Hands-on session 3 - (Building protein-ligand complexes containing non-standard residues with Antechamber) (H) | RW/AG/YM/YX/XW |
| 15:30 - 16:15 | Designing a good simulation project (L) | RW |
| 16:15 - 17:00 | What if there is no crystal structure? (L) | RW |
Day 3 (Wednesday 24th Aug 2011)
| 8:30 - 9:15 | Practical Guide to Running MM/PBSA calculations (L) | AG |
| 9:15 - 10:45 | Hands-on session 4 - (Introduction to binding energy calculations using MM-PBSA) (H) | RW/AG/YM/YX/XW |
| 10:45 - 11:15 | Coffee Break. | |
| 11:15 - 12:00 | Hands-on session 4 - (Introduction to binding energy calculations using MM-PBSA) Contd (H) | RW/AG/YM/YX/XW |
| 12:00 - 12:15 | Group Picture. | |
| 12:15 - 13:30 | Lunch. | |
| 13:30 - 14:20 | Statistical Mechanics for Free Energy Calculations (L) | AG |
| 14:20 - 17:00 | Hands-on session 5 - (Calculating relative binding free energies for protein/carbohydrate complexes) (H) | RW/AG/YM/YX/XW |
Day 4 (Thursday 25th Aug 2011)
| 8:30 - 9:15 | An overview of enhanced sampling techniques (L) | AG |
| 9:15 - 9:45 | Practical considerations for running replica exchange calculations (L) | RW |
| 9:45 - 10:45 | Hands-on session 6 - (Replica exchange simulations) (H) | RW/AG/YM/YX/XW |
| 10:45 - 11:15 | Coffee Break. | |
| 11:15 - 12:00 | Hands-on session 6 - (Replica exchange simulations) Contd (H) | RW/AG/YM/YX/XW |
| 12:00 - 13:30 | Lunch | |
| 13:30 - 15:15 | Hands-on session 6 - (Replica exchange simulations) Contd (H) | RW/AG/YM/YX/XW |
| 15:15 - 15:45 | Guest Lecture - Allan Hou [NVIDIA Inc.] (L) | AH |
| 15:45 - 16:30 | Considerations for Maximizing Performance (Parallel Execution and NVIDIA GPU Acceleration) (L) | RW |
| 16:30 - 17:00 | Practical considerations for running umbrella sampling calculations (L) | AG |
| 18:00 - 22:00 | Workshop Social / Dinner | |
| Park Café, Shanghai Marriott Hotel Changfeng Park, 158 Da Du He Road, Putuo District |
Day 5 (Friday 26th Aug 2011)
| 9:30 - 10:45 | Hands-on session 7 - (Umbrella sampling simulations) (H) | RW/AG/YM/YX/XW |
| 10:45 - 11:15 | Coffee Break. | |
| 11:15 - 12:00 | Hands-on session 7 - (Umbrella sampling simulations) Contd (H) | RW/AG/YM/YX/XW |
| 12:00 - 13:30 | Lunch | |
| 13:30 - 14:15 | An Introduction to QM/MM Calculations (L) | AG |
| 14:15 - 16:30 | Hands-on session 8 - (QM/MM MD Simulations) (H) | RW/AG/YM/YX/XW |
| 16:30 - 17:00 | Final Remarks / Question and Answer Session | RW/AG |
List of covered topics
- Introduction to force fields and molecular dynamics (Lecture)
- Overview of AMBER and AmberTools and its programs (Lecture)
- Introduction to setting up and running simulations (Lecture / Hands on)
- Visualizing AMBER simulations (Hands on)
- Overview of AMBER Force Fiels / Solvent Models etc. (Lecture)
- Introduction to implicit solvent and binding energy calculations (Lecture)
- Protein folding and advanced analysis (Hands on)
- Designing good simulation projects (Lecture)
- Dealing with non-standard residues (Lecture / Hands on)
- What to do if there is no crystal structure (Lecture)
- Statistical mechanics for free energy calculations (Lecture / Hands on)
- QM/MM coupled potential simulations (Lecture / Hands on)
- GPU accelerated molecular dynamics simulations (Lecture)
The following material is a non-exhaustive, fairly subjective list of information about the AMBER programs and computational chemistry in general. It is not a prerequisite for workshop participants but shall serve as a pointer for the interested.
- AMBER 11 manual
- AmberTools manual
-
D.A. Case, T.E. Cheatham, III, T. Darden, H. Gohlke, R. Luo, K.M. Merz, Jr., A. Onufriev, C. Simmerling, B. Wang and R. Woods. "The AMBER biomolecular simulation programs." J. Comput. Chem. 26 (2005) 1668-1688.
-
J.W. Ponder and D.A. Case. "Force fields for protein simulations." Adv. Prot. Chem. 66 (2003) 27-85.
-
O.M. Becker, A.D. MacKerrell, Jr., B. Roux, M. Watanabe, eds, "Computational Chemistry and Biophysics", Marcel Dekker, 2001: Chapter 3 "Dynamics Methods" pp39-67.
-
O.M. Becker, A.D. MacKerrell, Jr., B. Roux, M. Watanabe, eds, "Computational Chemistry and Biophysics", Marcel Dekker, 2001: Chapter 9 "Free Energy Calculations" pp169-197.
-
Andrew R. Leach, "Molecular Modelling - Principles and Applications", 2nd Edition (2001), Prentice Hall, Harlow, UK.
-
Daan Frenkel and Berend Smit, "Understanding Molecular Simulation" 2nd Edition (2001), Academic Press, London, UK.
-
Frank Jensen, "Introduction to Computational Chemistry", 2nd Edition (2006), Wiley VCH, Chichester, UK.

{kind=link}